
关于大样本量差异分析是否可以转为拟时序分析,以及两个分组的差异分析仅仅是上下调的问题,以下是经过伪原创处理后的文章:
许多朋友在后台表示对单细胞数据分析中的拟时序分析感到困惑。恰逢最近看到了一篇清晰展现拟时序分析重要性的文献,这里与大家分享。这篇文献完美地展示了为什么单纯的差异分析不足以揭示全部信息,而拟时序分析则是对差异分析的深入剖析。
这篇发表在NATURE COMMUNICATIONS | (2021) 的文章:《CD177 modulates the function and homeostasis of tumor-infiltrating regulatory T cells》,链接是:https://www.php.cn/link/1ee9bee2c7227c35ac1ca90f2e4fb172:
研究开始时使用了13,433个外周血单核细胞(PB)和12,239个肿瘤浸润细胞(TI),通过降维聚类分群后,根据FOXP3和CD25 (IL2RA)定位到了Treg亚群,分别是160个PB和574个TI Treg细胞。
 降维聚类分群
降维聚类分群
如果直接对这两个分组进行差异分析,可以识别出273个差异表达基因(DEGs)(Log fold-change > 1, adjusted p-value < 0.05)。
此外,作者在自己的ccRCC单细胞矩阵以及一个公共数据集HCC中也进行了类似的差异分析,并筛选出共有基因:
 差异基因及其交集
差异基因及其交集
这样的差异分析虽然进行了交集筛选,但仍遗漏了许多细节,仅得到基因的上下调信息,而忽略了每个基因在两个单细胞亚群中具体的渐变趋势。
由于细胞数量并不多,运行Monocle 2算法,可以清楚地看到上下调的变化趋势,甚至发现隐藏的变化模式:
首先是拟时序分析的轨迹流形图,展示了ccRCC中Treg细胞的轨迹,使用Monocle 2算法。实线和虚线代表由表达谱定义的不同细胞轨迹/命运。
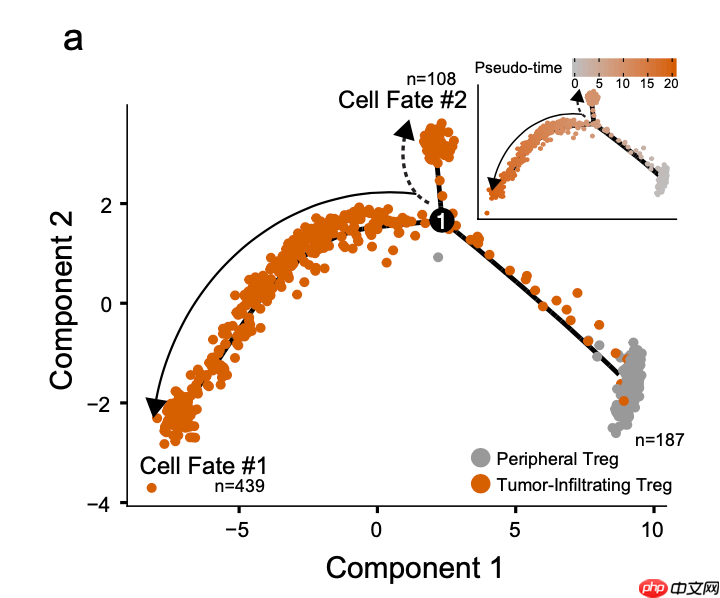 拟时序分析结果
拟时序分析结果
虽然拟时序分析只展示了上述的一个图,但具体的代码步骤还是有一定难度的。以下是可直接使用的代码示例,我们以SeuratData包中的pbmc3k数据集为例,主要是将Seurat包的对象转换为monocle中的单细胞对象:
代码语言:javascript
library(SeuratData) # 加载seurat数据集
getOption('timeout')
options(timeout=10000)
#InstallData("pbmc3k")
data("pbmc3k")
sce <- as.SingleCellExperiment(pbmc3k[, 1:10, ])
sc_cds <- importCDS(sce, import_all = TRUE)
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
cds <- detectGenes(cds, min_expr = 0.1)
cds <- setOrderingFilter(cds, ordering_genes = row.names(subset(fData(cds), num_cells_expressed >= 10)))
cds <- reduceDimension(cds, max_components = 2, method = 'DDRTree')
cds <- orderCells(cds)
save(cds, file = 'input_cds.Rdata')确保前面的步骤无误后,接下来就可以运行拟时序分析的主程序:
代码语言:javascript
rm(list=ls()) options(stringsAsFactors = F) library(monocle) library(Seurat) load(file = 'input_cds.Rdata') # 接下来很重要,到底是看哪个性状的轨迹 colnames(pData(cds)) table(pData(cds)$Cluster) table(pData(cds)$Cluster, pData(cds)$celltype) plot_cell_clusters(cds, 1, 2) ## 我们这里并不能使用 monocle的分群 # 还是依据前面的 seurat分群, 其实取决于自己真实的生物学意图 pData(cds)$Cluster = pData(cds)$celltype table(pData(cds)$Cluster) Sys.time() diff_test_res <- differentialGeneTest(cds, fullModelFormulaStr = "~Cluster") save(diff_test_res, cds, file = 'output_of_phe2_monocle.Rdata')
有了上面的output_of_phe2_monocle.Rdata文件后,就是拟时序分析的结果,接下来就可以进行各种可视化。首先看看每个细胞的时序信息:
LimeSurvey是一款在线问卷管理系统,具有问卷的设计、修改、发布、回收和统计等多项功能。同时它也是一个开源软件,其最新版本的软件包可以完全免费获取和使用。它集成了调查程序开发、调查问卷的发布以及数据收集等功能,使用它,用户不必了解这些功能的编程细节。 网上收集的调查数据可以导出多种文件格式以便分析,例如 spss数据格式 *.dat文件。
 198
198

代码语言:javascript
## 后面是对前面的结果进行精雕细琢
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(gplots)
library(ggplot2)
library(monocle)
library(dplyr)
load(file = 'output_of_phe2_monocle.Rdata')
cds = my_cds_subset
colnames(pData(cds))
table(pData(cds)$State, pData(cds)$Cluster)
library(ggsci)
p1 = plot_cell_trajectory(cds, color_by = "Cluster") + scale_color_nejm()
p1
ggsave('trajectory_by_cluster.pdf')
plot_cell_trajectory(cds, color_by = "celltype")
p2 = plot_cell_trajectory(cds, color_by = "Pseudotime")
p2
ggsave('trajectory_by_Pseudotime.pdf')
p3 = plot_cell_trajectory(cds, color_by = "State") + scale_color_npg()
p3
ggsave('trajectory_by_State.pdf')
library(patchwork)
p1 + p2 / p3可视化结果如下:
 拟时序分析的多元化结果
拟时序分析的多元化结果
因为原文中恰好形成了两个细胞命运,所以可以使用BEAM函数,仍然是提取具体的基因进行热图可视化:
b 基于流形的免疫基因转录变化的拟时序投影。显著性基于相对于起始位置的差异测试,该起始位置也用于生成拟时序并进行了多重比较调整。
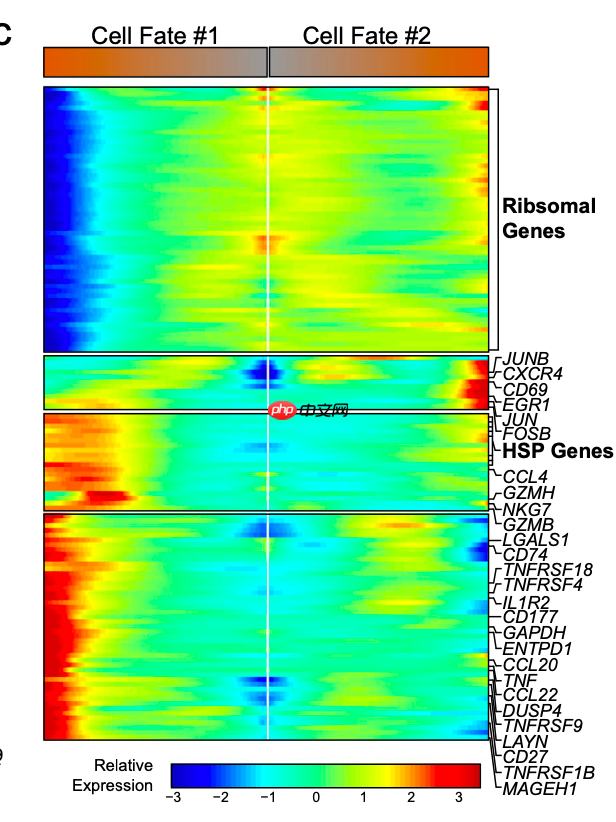 拟时序分析的差异基因热图
拟时序分析的差异基因热图
热图中的基因有多种展示方式:
c 显著基因(q < 0.05)的表达热图
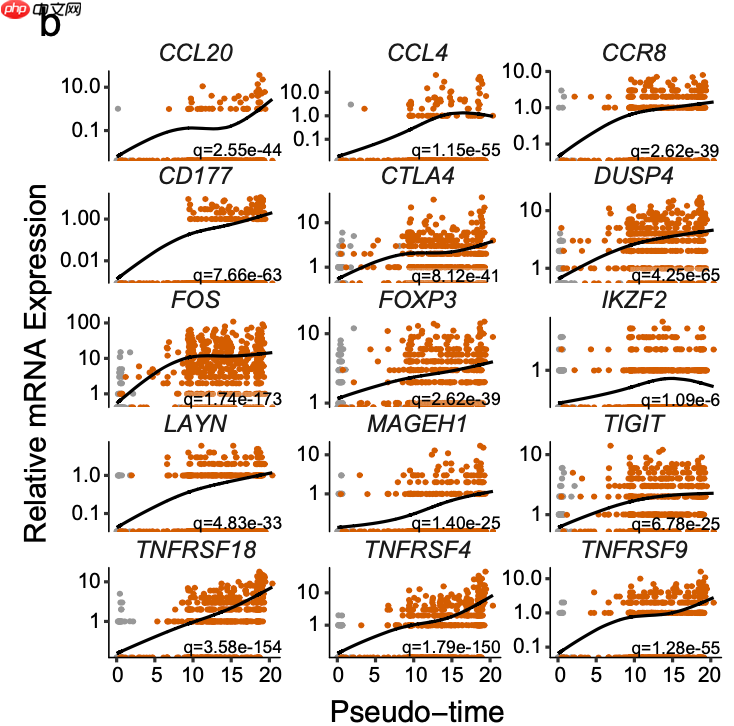 拟时序分析的差异基因趋势折线图
拟时序分析的差异基因趋势折线图
以及:
d 基于流形的免疫基因转录变化的细胞轨迹投影。显著性基于TI Treg细胞第一和第二细胞命运之间的差异测试。“x”表示流形两极的标尺化平均mRNA水平。
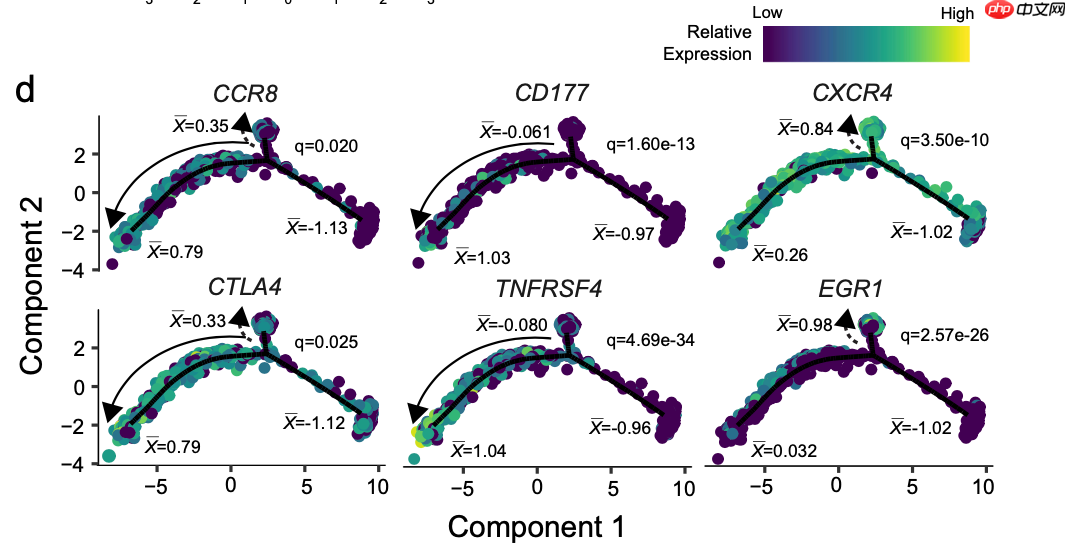 拟时序分析的差异基因表达量图
拟时序分析的差异基因表达量图
最后这个图,看起来技术含量十足!
通过以上内容可以看出,拟时序分析不仅可以揭示差异分析无法捕捉的动态变化,还能提供更细致的基因表达趋势和细胞命运信息。因此,大样本量差异分析转为拟时序分析可以提供更深入的洞察,而两个分组的差异分析不仅仅是简单地识别上下调基因,更重要的是理解这些变化的动态过程。
以上就是拟时序分析就是差异分析的细节剖析的详细内容,更多请关注php中文网其它相关文章!

每个人都需要一台速度更快、更稳定的 PC。随着时间的推移,垃圾文件、旧注册表数据和不必要的后台进程会占用资源并降低性能。幸运的是,许多工具可以让 Windows 保持平稳运行。

Copyright 2014-2025 https://www.php.cn/ All Rights Reserved | php.cn | 湘ICP备2023035733号





 430
430























